Regulation of actin filament length is a central process by which eukaryotic cells control the shape, architecture, and dynamics of their actin networks. This regulation plays a fundamental role in cell motility, morphogenesis, and a host of processes specific to particular cell types. This paper by recently graduated [Biophysics and Structural Biology] Ph.D. student Jeffrey Bombardier and collaborators resolves the long-standing mystery of how formins and capping protein work in concert and antagonistically to control actin filament length. Bombardier used the CoSMoS multi-wavelength single-molecule fluorescence microscopy technique to to discover and characterize a novel tripartite complex formed by a formin, capping protein, and the actin filament barbed end. Quantitative analysis of the kinetic mechanism showed that this complex is the essential intermediate and decision point in converting a growing formin-bound filament into a static capping protein-bound filament, and the reverse. Interestingly, the authors show that “mDia1 displaced from the barbed end by CP can randomly slide along the filament and later return to the barbed end to re-form the complex.” The results define the essential features of the molecular mechanism of filament length regulation by formin and capping protein; this mechanism predicts several new ways by which cells are likely to couple upstream regulatory inputs to filament length control.
Single-molecule visualization of a formin-capping protein ‘decision complex’ at the actin filament barbed end
Jeffrey P. Bombardier, Julian A. Eskin, Richa Jaiswal, Ivan R. Corrêa, Jr., Ming-Qun Xu, Bruce L. Goode, and Jeff Gelles Nature Communications 6:8707 (2015)
The capping protein expression plasmid described in this article is available from Addgene.
Readers interested in this subject should also see a related article by Shekhar et al published simultaneously in the same journal. We are grateful to the authors of that article for coordinating submission so that the two articles were published together.
Cells contain thousands of protein “micromachines” performing a bewildering number of chemical reactions every second. The challenge for biologists in the 21st century is to integrate information about multiple – or even all – proteins into holistic models for the entire cell. This is a daunting task. The addition of any new component to a system can alter the behavior of the components already there. This phenomenon is especially familiar to biologists studying the cytoskeleton, a complex system of protein filaments that provide the force for cell division and migration, among other things. The building blocks of the cytoskeleton are simple proteins called tubulin and actin that assemble into a remarkable variety of shapes depending on context. While the basic chemistry of this assembly process has been understood in purified systems for decades, how it happens in cells is not well understood. For example, growth of actin filaments is a two-step process: nucleation, or the formation of a new filament, and elongation, or the extension of that existing filament. Both steps happen just fine when actin is present in pure form in a test tube. In cells, however, proteins called profilin and capping protein block these two steps, respectively. Nucleation and elongation can only occur because other proteins overcome these blocks. Thus, a faithful experimental system to study actin assembly as it would occur in a living cell requires – at a minimum – five purified proteins.
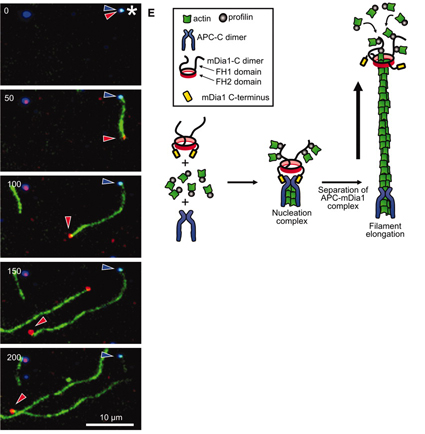
One technological advance of great importance is the ability to literally see single molecules (in this case proteins) using advanced fluorescence imaging. In such an experimental system, many details can be captured. In a recent publication in Science, Dr. Dennis Breitsprecher and colleagues in the Goode and Gelles labs, undertook this challenge and directly visualized the effects of key regulatory proteins helping actin proteins nucleate and grow into filaments in the presence of both profilin and capping protein. A previous study from the Goode lab had shown that two proteins, called APC and mDia1, together stimulate the growth of actin into filaments (Okada et al, 2010). In the present study, Breitsprecher and colleagues examined the mechanism by which APC and mDia1 overcome the profilin and capping protein-imposed blocks. To do this, they ‘tagged’ actin, APC and mDia1 with three different fluorescent dyes, each of a different color, and then filmed these molecules (using triple-color TIRF microscopy) in the act of building an actin filament to learn precisely what they are doing.
The authors began by imaging APC and actin (2 colors) at the same time. APC formed discrete spots on the microscope slide, and growing actin filaments emerged from them, suggesting than APC nucleates actin filament formation. As the filament emerged from the APC spot, APC stayed where it was: remaining stably associated at the site of nucleation. Next, the authors added dye-labeled mDia1 to the system, and observed mDia1 molecules staying attached to and ‘riding’ the fast-growing end of actin filament, while protecting it from capping protein.
The most remarkable result came when they visualized all three labeled molecules together (actin, APC, and mDia1). What they saw was that APC and mDia1 first come together in a stable complex even before actin arrives. Then APC recruits multiple actin subunits to initiate the nucleation of a filament. This complex was resistant to the blocks imposed by both profilin and capping protein. As the filament grew from the APC-mDia1 spot, mDia1 separated from APC and stayed bound to the growing end of the filament – protecting it from capping protein while it grows. Thus, even though APC and mDia1 have different activities, participating in different stages of the growth of a filament, they associate together before actin even arrives, likely so that once the actin filament is born, it is immediately protected from capping protein. This mechanism has been compared to a rocket launcher: APC is the launch pad for an actin filament, which is then propelled forward by mDia1.
Rocket launcher mechanism for APC and mDia1 nucleation. Left: Microscopic image of a growing actin filament. APC stays put while mDia1 remains associated with the fast growing end. Right: Model for the rocket launcher mechanism.
The new study provides great detail of the system: for example, the number of APC subunits required to nucleate actin filaments was determined, and the growth rate of actin filaments in the presence and absence of all the other components was measured. Ultimately, all of these data will be required to put together a detailed model of how actin filaments grow inside of real cells: details that would be difficult or impossible to obtain without employing single molecule analysis.
For the future, the authors have set their sights on even more challenging experiments aimed at elucidating the mysterious link between tubulin and actin fibers. APC and mDia1 are implicated in this linkage in living cells, but almost nothing is known about how they physically link and/or communicate information between the two systems. Since APC is mutated in some 80% of colon cancer tumors, understanding its multiple roles is of clinical as well as intellectual importance. This will be an exciting, if challenging, endeavor for the future.
Some video resources if you need to explain scientific topics to students (or need something explained to you!)
iBioMagazine.org features short (<15 min) talks that highlight the human side of research. iBioSeminars.org provides approximately hour-long seminars by high profile researchers.
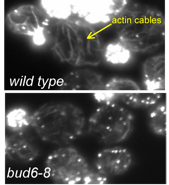
Formins are a family of proteins conserved across a wide range of eukaryotes and constitute a major class of actin nucleators. In a paper recently published in Molecular Biology of the Cell, a team led by Ph.D. student Brian Graziano in the laboratory of Professor Bruce Goode made the surprising finding that formins depend on co-factors to efficiently nucleate actin assembly both in vitro and in vivo. This discovery was unanticipated because earlier studies had shown that purified formins are sufficient to catalyze actin polymerization in vitro. Graziano, working in collaboration with the labs of Laurent Blanchoin and Isabelle Sagot, investigated the mechanism and function of a formin-binding protein called Bud6 and found that it elevates formin nucleation activity by 5-10 fold. Further, they showed that this activity of Bud6 is critical in vivo for maintaining normal levels of actin cable assembly and polarized cell growth (see figure).
Earlier work from the Goode lab had shown that Bud6 enhances formin-mediated actin assembly in vitro (Moseley et al., 2004), but had left open the question of whether Bud6 stimulates the nucleation or elongation phase of filament growth (an important mechanistic distinction), and whether the activities of Bud6 are important in vivo. Graziano and collaborators dissected Bud6 mechanism by: (a) generating mutations in Bud6 that separately disrupt its interactions with formins (bu6-35) and actin monomers (bud6-8), (b) using TIRF (total internal reflection fluorescence) microscopy to visualize the effects of Bud6 and formins on individual actin filaments polymerizing in real time, and (c) performing a genetic analysis of bud6 alleles. They made three important observations. First, Bud6 enhances the nucleation rather than elongation phase of actin assembly, in sharp contrast to another formin ligand, profilin, that enhances elongation. Second, this activity of Bud6 requires its direct interactions with both the formin and actin monomers, suggesting that Bud6 recruits monomers to the formin to help assemble an actin ‘seed’. Third, genetic perturbation of these activities of Bud6 results in reduced levels of actin cable formation in vivo, in turn causing defects in polarized secretion and cell growth.
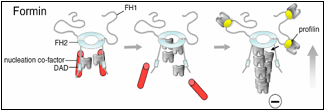
Until now, formins were thought to nucleate actin assembly by themselves, which is mechanistically distinct from the Arp2/3 complex (another major actin nucleator). Efficient nucleation by Arp2/3 requires the addition of a nucleation-promoting factor (NPF) such as WASp or WAVE, which recruits actin monomers. Graziano et al. reveal that some formins are similar to Arp2/3 in that they too require an NPF for robust nucleation. Their findings also uncover unanticipated mechanistic parallels between the two systems, since in each case nucleation requires both an actin filament end-capping component (formin or Arp2/3) and an actin monomer-recruiting factor (Bud6 or WASp).
How well is this formin-NPF mechanism conserved? Clues to this question have recently emerged from other studies. A paper published last year in The Journal of Cell Biology by the Goode lab, working in collaboration with the labs of Niko Grigorieff (Brandeis) and Gregg Gundersen (Columbia), implicates the human tumor suppressor protein Adenomatous polyposis coli (APC) in functioning as a formin NPF (Okada et al., 2010). Another study published in The Proceedings of the National Academy of Sciences by the labs of Mike Eck (Dana Farber Cancer Institute), Margot Quinlan (UCLA), and Avital Rodal (Brandeis), suggests that Spire, which is conserved in mammals and flies, may serve as a formin NPF (Vizcarra et al., 2011). Bud6, Spire, and APC all bind multiple actin monomers and interact with the C-terminus of formins to enhance actin assembly, suggesting that they may have related mechanisms and perform functionally analogous roles.
Although the requirement of NPFs increases the complexity of the formin mechanism, it offers an explanation for how cells simultaneously overcome two prominent barriers to actin assembly found in vivo – actin monomer binding proteins (e.g. profilin) that suppress formation of an actin nucleus and capping proteins that terminate growth by associating with the growing end of the filament. NPFs can facilitate nucleation by recruiting actin monomers in the presence of profilin, and formins protect growing ends of filaments from capping proteins. Future work will focus on identifying new formin-NFP pairs, defining the cellular processes with which they are associated, and distinguishing the underlying mechanistic differences among each set.
When large structures are built inside of cells, how are their dimensions determined? Are cues received that tell the structure to keep growing, or to slow down, or to stop growing altogether? A recent study published in Developmental Cell by a team led by Molecular and Cell Biology PhD student Melissa Chesarone-Cataldo and Professor of Biology Bruce Goode begins to address these questions by focusing on cytoskeletal structures called yeast actin cables.
Actin cables serve as essential railways for myosin-dependent transport of vesicles, organelles and other cargo, required for yeast cells to grow asymmetrically and produce a daughter cell. Cables are assembled at one end of the mother cell and run the length of the entire cell, but no longer, or else they would hit the back of the cell, buckle and misdirect transport. So how does an actin cable know how long to grow? How are other properties of the cable, such as its thickness and mechanical rigidity determined, and how important are these properties for cable function in vivo?
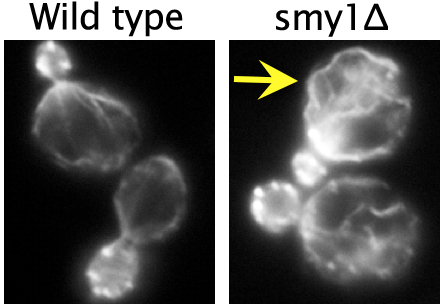
Actin cables are assembled at the bud neck by the formin protein Bnr1, and rapidly extend into the mother cell at a rate of 0.5-1 µm/s. At this speed, the tip of the actin cable reaches the back end of the cell in about 5-10 seconds. Each cable consists of many shorter overlapping pieces (individual actin filaments) that are stitched or cross-linked together to form a single cable, and cables continuously stream out of the bud neck due to the robust actin assembly activity of Bnr1. Chesarone-Cataldo et al. asked the question, “what mechanism prevents the cables from colliding with the back of the cell and overgrowing?” In doing so, they identified a novel actin cable ‘length sensing’ feedback loop, dependent on the myosin-passenger protein Smy1.
Using live-cell imaging, they showed that Smy1 molecules are transported by myosin from the mother cell to the bud neck, where they pause to interact with the formin Bnr1. Purified Smy1 attenuated Bnr1 activity by slowing down the rate of actin filament elongation. When the SMY1 gene was deleted, cables grew too long, hit the rear of the cell and buckled (see image, right). In addition, the mutant cables abnormally fluctuated in thickness and were kinked, impairing transport of myosin and its cargoes.
The authors propose that a negative feedback loop controls actin cable length. In their model, the cargo (Smy1 in this case) communicates with the machinery that is making the cable (the formin Bnr1), as a means of sensing ‘railway’ length. The longer the railway grows, the more passengers it picks up, and the more transient inhibitory pulses the formin receives. As such, longer cables are selectively attenuated, while shorter cables are allowed to grow rapidly. This negative feedback loop allows yeast cells to tailor actin cable length to the dimensions of the cell and to the needs of its myosin-based transport system.
Current work in the Goode lab is aimed at testing many of the mechanistic predictions of the model above and understanding how Smy1 functions in coordination with other known regulators of Bnr1, all simultaneously present in a cell, to produce actin cables with proper architecture and function. In addition, experiments are underway to find out whether related mechanisms are used to control formins in mammalian cells and to understand the physiological consequences of disrupting those mechanisms.
Chesarone-Cataldo M, Guérin C, Yu JH, Wedlich-Söldner R, Blanchoin L, Goode BL. The Myosin passenger protein Smy1 controls actin cable structure and dynamics by acting as a formin damper. Dev Cell. 2011 Aug 16;21(2):217-30.
A new review article in Current Opinion in Cell Biology by Molecular and Cell Biology grad student Melissa Chesarone and Biology’s Professor Bruce Goode focuses on a group of remarkable protein machines that rapidly assemble actin polymers in cells. These factors are essential for cell division, cell movement, and cell shape determination in virtually all organisms. Their catalytic mechanisms involve intricate fast-moving parts, which enables them to construct entire actin networks in a matter of seconds.
Single particle electron microscopy reconstruction can be a powerful tool for determining the structure of large protein complexes. One limitation of the technique is the difficulty in coming up with specific labels for the protein that can be visualized with EM. In a new paper in RNA, postdoc Beth Stroupe and coworkers show that the use of the actin-nucleating protein Spire as a cloneable tag allows them to nucleate actin filaments that then “point” to the location of the tag in the complex seen in EM, and applied the technique to their studies of the C complex spliceosome.